|
1 IVITA, Facultad de Medicina Veterinaria, Universidad San Marcos, Lima, Perú. Correo electrónico: rrosadio@computextos.com.pe
2 CONOPA
3 Consejo Nacional de Camélidos Sudamericanos, CONACS, Lima, Perú. Correo electrónico: conacs@amauta.rcp.net.pe
4 Conservation Genetics Group, Institute of Zoology, Regent’s Park, London, U.K. Correo electrónico: Miranda.Kadwell@UKgateway.net
5 Cardiff School of Biosciences, Cardiff University, Cardiff, U.K. Correo electrónico: BrufordMW@Cardiff.ac.uk
OBJETIVOS DEL PROYECTO
El objetivo científico de este proyecto (financiado por la Iniciativa Darwin para la Sobrevivencia de Especies, Departamento del Medio Ambiente, Reino Unido), fue usar marcadores genéticos moleculares, específicamente microsatélites, para:
- elucidar la reciente historia evolutiva de poblaciones silvestres de los camélidos peruanos;
- evaluar la presente diversidad genética y su distribución dentro de estas poblaciones;
- identificar “unidades de manejo” demográficamente independientes dentro de estas poblaciones para manejo futuro
- estimar el impacto genético producido por estrategias de manejo pasadas y futuras, incluyendo las consecuencias del programa de utilización sostenible.
Es importante enfatizar que este es el primer estudio de su clase llevado a cabo en camélidos sudamericanos silvestres.
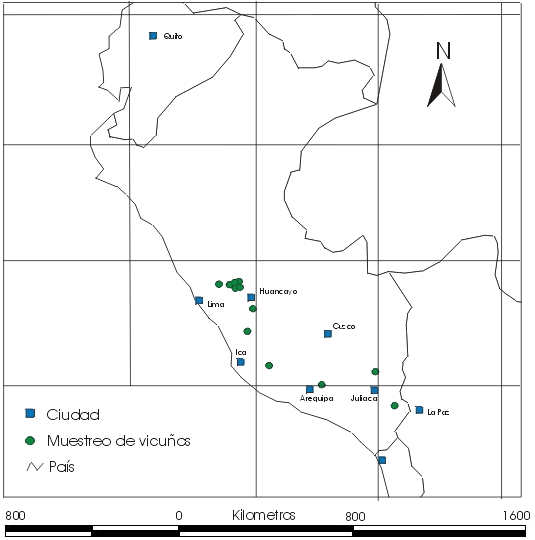
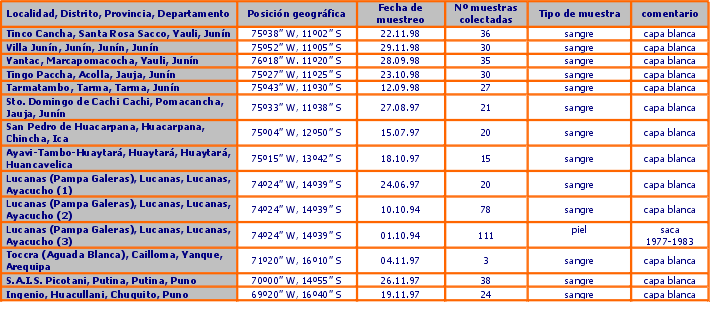
Se muestrearon 12 poblaciones (Figura 1, Tabla 1) seleccionadas por las siguientes razones:
- Por su ubicación en puntos geográficos dispersos a través de la distribución de vicuñas en el Perú ;
- Por su historia de aislamiento geográfico; y
- Por aparentemente no haber sufrido la introducción de animales procedentes de la Reserva de Pampa Galeras.
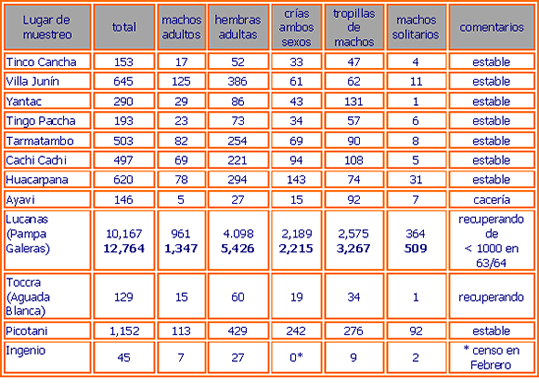
En la tabla 1 se presenta información sobre la ubicación geográfica de las poblaciones muestreadas, el número de muestras tomadas y detalles adicionales. Las muestras de sangre y piel fueron subdivididas en dos partes, la mitad se guardó en la Unidad de Virología y Genética Molecular FMV - UNMSM y la otra mitad fue enviada al Instituto de Zoología de Londres con los permisos de exportación CITES/Perú 00240 y 00658, y permisos de importación UK/EU CITES 67264 y 206242/01. En la tabla 2 se presenta detalles de las poblaciones muestreadas según los censos de CONACS realizados en 1997 y 1999, y la composición demográfica de cada población. También se anota cualquier evidencia anecdótica de la historia demográfica reciente.
Se tomó muestras de sangre (en tubos vacutainer con EDTA) o de piel. Luego en el laboratorio se extrajo el ADN usando métodos convencionales que incluyen la digestión del material celular usando Proteinasa K seguido de la extracción de ácido nucleico usando fenol y fenol/cloroformo para remover las proteínas y finalmente precipitar el ADN con etanol 100% (para más detalles ver Bruford et al., 1998). El ADN fue resuspendido en una solución de buffer TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) hasta su análisis.
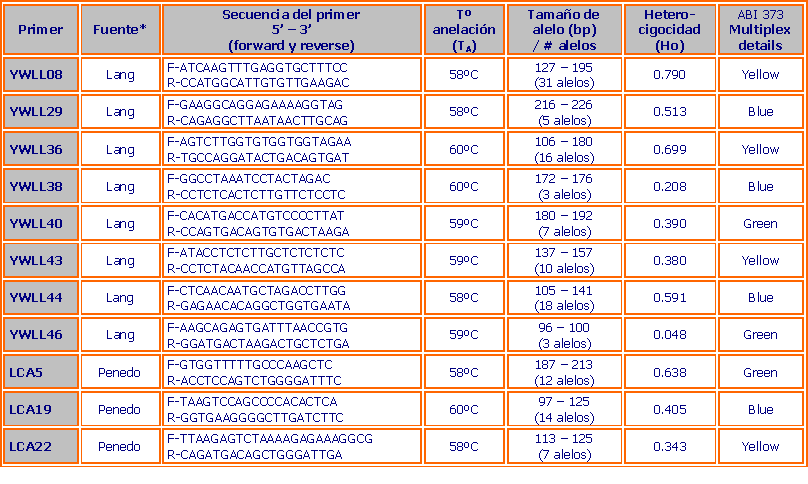
Se analizaron 11 microsatélites para camélidos sudamericanos, publicados previamente (Lang et al., 1996; Penedo et al., 1998). Los microsatélites son marcadores nucleares (ADN nuclear) encontrados en los cromosomas de la mayoría de los eucariotas, en muchas especies son altamente polimórficos por lo que se emplean para pruebas de paternidad, perfiles individuales en forensia, análisis de poblaciones y estudios de hibridización (Goldstein y Schlötterer, 1999). Previamente, nosotros demostramos que los microsatélites pueden ser usados efectivamente en estudios de hibridización en camélidos sudamericanos (Kadwell et al., en revisión) y aquí se utilizaron estos marcadores para medir la diversidad genética de las poblaciones e inferir su historia demográfica reciente.
En la tabla 3 se presentan detalles de los microsatélites usados, su diversidad genética en términos de heterocigocidad (la proporción de individuos que heredan diferentes alelos para un locus determinado o gen de sus padres, este es un buen indicador de variación genética) y tamaño de alelos en vicuñas peruanas. En la tabla 4 se presenta los resultados del análisis de diversidad genética dentro de cada población comparando la heterocigocidad observada y la heterocigocidad esperada (asumiendo cruces al azar con respecto a la similaridad genética de los individuos) y número promedio de alelos por locus. Una gran diferencia entre heterocigocidad observada y esperada puede ser causada por cruces no al azar (consanguinidad) o selección que actúa contra ciertos genotipos.
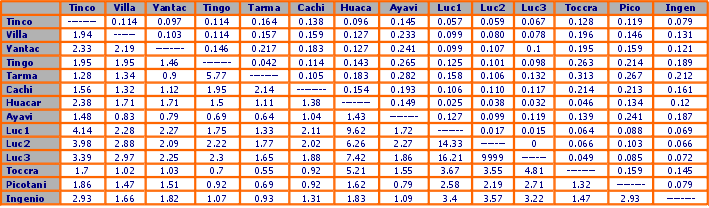
En la tabla 5 se indica los valores pareados de la diversidad genética entre poblaciones, medidas por el comúnmente usado índice FST que varía entre cero (no hay diferencia genética) y uno (diferencia completa). Se puede interpretar también como el componente de la variación genética total entre dos poblaciones que se puede atribuir a las diferencias entre ellas. FST se calcula como:
FST = HT – HS/ HT
donde HT es la heterocigocidad esperada en la población total y HS es la heterocigocidad esperada en la subpoblación estudiada. La tabla 5 también incluye los valores Nm (número estimado de migrantes por generación) que se calcula de FST usando la fórmula:
Nm = (1-FST)/4FST
Nm se incluye para efectos de ilustración, debido a que implica asunciones acerca de las poblaciones que no necesariamente se cumplen en la mayoría de los casos. Todos los cálculos y procedimientos de análisis se efectuaron empleando los programas GENEPOP v3.1(Raymond y Rousset, 1995) y GENETIX v4.0 (Belkhir, 1999).
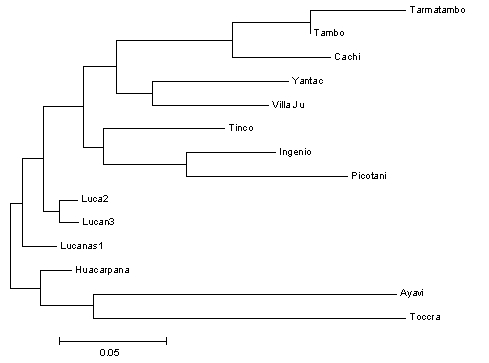
La figura 2 muestra un dendograma (árbol genealógico de poblaciones), creado usando el análisis “neighbour-joining” (agrupando vecinos) (Saitou et al., 1987) en el programa para computadora MEGA (Kumar et al., 1993), que agrupa poblaciones de acuerdo a la distancia genética (Nei, 1972) (análisis de variación genética en forma análoga a FST) que permite identificar agrupaciones de poblaciones relacionadas.
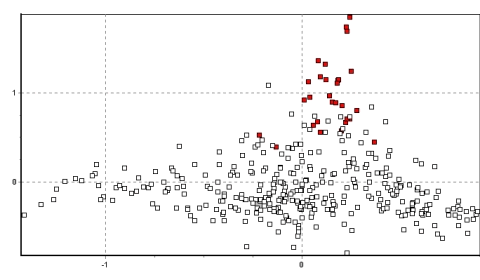
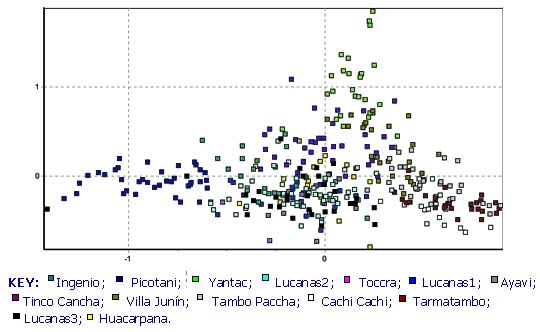
La figura 3 muestra un plot bidimensional de correspondencia factorial de todas las poblaciones, donde la diversidad genética entre poblaciones es expresada como factores que explican la correspondencia entre muestras en un número de dimensiones (aquí mostramos las dos dimensiones que explican la más alta proporción de correspondencia; Benzécri, 1973 usando GENETIX v. 4.0 - para una explicación más completa ver resultados). Las relaciones espaciales entre poblaciones pueden ser adjudicadas examinando la manera como los individuos de cada población se agrupan en 2, 3 ó más dimensiones. Las figuras 4a,b,c,d muestran respectivamente las localizaciones aproximadas de los cuatro principales grupos poblacionales identificados por este método : Junín Sur, Junín Noroeste, Andes centrales (Huancavelica-Arequipa) y Puno.
La tabla 3 muestra claramente que los marcadores usados son altamente polimórficos e informativos en las vicuñas peruanas, lo que significa que estos marcadores son ideales para el estudio de diversidad genética y diferenciación. Por ejemplo, YWLL08 es excepcionalmente informativo, posee 31 alelos y cerca del 80% de los individuos son heterocigotes. Interesantemente, nuestra investigación ha encontrado más alelos de los previamente reportados en los estudios iniciales por los investigadores pioneros.
Por ejemplo, Lang et al. (1996), aislaron los marcadores YWLL usados en este estudio, en ADN de llamas, identificando 13 alelos para YWLL08 y 6 alelos para YWLL36 (comparado con los 31 y 16 encontrados en este estudio respectivamente). Por otro lado, Penedo et al. (1998) aislaron los microsatélites LCA usados en nuestro estudio, también en ADN de llama y encontraron solamente 7 alelos para LCA5 y 10 alelos para LCA22 (comparados con los 12 y 14 encontrados en nuestro estudio respectivamente). Sin embargo, los niveles de heterocigocidad son mucho más bajos en este estudio comparado con los reportes originales.
Quizás esto sea sorprendente, debido a que se ha muestreado poblaciones silvestres (vicuñas) y luego fueron comparadas con los camélidos domésticos. Este resultado, probablemente se debe a que las vicuñas peruanas tengan una variación genética más baja dentro de sus poblaciones, aunque esto pareciera ilógico, dado que la vicuña posee muchos más alelos que las muestras de camélidos domésticos. Sin embargo, esto podría ser explicado si la mayoría de la diversidad genética en vicuñas peruanas se encontrara entre poblaciones en lugar de dentro de poblaciones.
Una explicación alternativa es el fenómeno conocido como “ascertainment bias”. Al usarse microsatélites desarrollados y escogidos por ser altamente polimórficos en llamas es probable que estos sean menos variables en vicuñas. Además nuestros estudios previos (Kadwell et al., en revisión) indican que las llamas domésticas derivan del guanaco sin ninguna injerencia de la vicuña. Es bastante común observar que los microsatélites aislados en una determinada especie (en este caso, llama) son menos variables en otras especies a pesar de cuan cercanamente relacionadas estas sean como es el caso de los camélidos sudamericanos. Sin embargo, hasta que no se disponga de datos equivalentes en guanaco será difícil estimar verdaderamente la contribución relativa de los dos potenciales factores explicativos, y es posible (tal vez cierto) que ambos puedan jugar un rol.
La tabla 4 muestra los niveles de heterocigocidad encontradas en todas las poblaciones analizadas. Los valores promedio de heterocigocidad esperada varían entre 0.377 (Tamatambo) y 0.586 (Lucanas 2) para todos los loci. Estos valores son ciertamente más bajos que los comúnmente encontrados en poblaciones de mamíferos continentales (datos no mostrados), los cuales usualmente no son inferiores de 0.5, lo que podría reflejar una historia reciente de aislamiento poblacional, con cambios genéticos producidos por “genetic drift” (demográficamente mediados por cambios al azar en la frecuencia de alelos los cuales pueden llevar a una uniformidad genética y/o consanguinidad) o puede ser a consecuencia del tipo de microsatélites usados (ver arriba).
Sin embargo, como este fenómeno se encuentra en muchos de los loci usados, el cambio genético (“genetic drift”) o consanguinidad parecían ser la explicación más probable. Esta hipótesis es apoyada por las observaciones mostradas en la tabla 4, en donde la heterocigocidad observada es menor que la heterocigocidad esperada (el valor esperado de las frecuencias alélicas observadas bajo la suposición de cruzamiento al azar) en todas las poblaciones excepto una (Toccra, el cual tiene un tamaño muestral muy pequeño y podría estar sesgado). La observación es consistente con intracruzamiento localizado o selección contra heterocigotes. Además, hay un exceso significativo de homocigocidad en todas las poblaciones excepto en Huacarpana (ver Poblaciones Anómalas abajo), Lucanas1, Cachi Cachi, Toccra y Tarmatambo (análisis GENEPOP), lo que sugiere que consanguinidad y/o “genetic drift” es más o menos común.
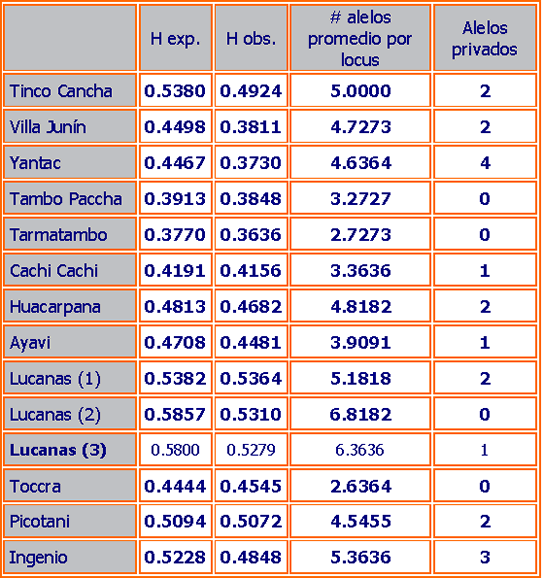
La tabla 4 también muestra la presencia de un total de 20 alelos privados (población-específico) que es más alto de lo esperado para una muestra en una población continental, e igualmente sugiere algún nivel de aislamiento local y cambio genético. Interesantemente, la frecuencia de alelos privados no parece corresponder con el tamaño de población o grado de aislamiento, sin embargo la población situada más al noroeste (Yantac) posee el más alto número de alelos únicos.
es el resultado del cruzamiento entre parientes y puede ser medido por un coeficiente de consanguinidad F, que varía entre 0 y 1, pero donde los valores encima de 0.05 son considerados altos. Por ejemplo, cruces hermano/hermana resultan en valores F de 0.25. Sin embargo, la consanguinidad en vicuñas, es altamente improbable bajo circunstancias normales debido a su sistema social, donde ambos sexos se dispersan de su grupo natal. Generalmente un elevado coeficiente de consanguinidad puede resultar de una situación donde una población ha atravesado por un cuello de botella demográfico resultado de su casi desaparición.
es el proceso en el cual frecuencias de alelos tienden a fluctuar ampliamente en pequeñas poblaciones debido a los efectos estocasticos de muestreo. Una vez que un alelo se ha perdido a través de cambio genético, sólo puede ser recuperado a través de mutaciones o flujo de genes. Así mismo, una vez que un alelo llega a fijarse en una población, todos los individuos son homocigotes para ese alelo a menos que mutación o inmigración introduzcan nuevas variantes.
Finalmente, aunque es claro que la diversidad alélica de la población total de vicuñas es alta en comparación con la encontrada en camélidos domésticos por Lang et al. 1996 y Penedo et al. 1998, el número de alelos por locus dentro de la población es menor de lo comúnmente observado en otros mamíferos (por ejemplo, Tarmatambo con 2.7 alelos por locus). Solamente Lucanas 2 y 3 muestran diversidad alélica relativamente alta (6.8 y 6.36 respectivamente), semejante a la frecuentemente encontrada en otros estudios.
Se calcularon índices de valores pareados de diferenciación genética entre poblaciones (FST Nei, 1972) con la finalidad de inferir relaciones entre poblaciones, para calcular las tasas (“rates”) de migración entre ellos y en combinación con los métodos de agrupamiento, para mostrar gráficamente los resultantes grupos demográficos. La Tabla 5 muestra valores FST pareados entre todas las poblaciones y los valores fluctúan desde cero en Lucanas 2 y 3 indicando completa similaridad genética (no es sorprendente desde que las muestras fueron tomadas de una misma población) y 0.31 entre Toccra y Tarmatambo, un alto grado de diferenciación genética entre dos poblaciones de mamíferos continentales. En general los valores FST son altos, con 67% excediendo 0.1, todas las comparaciones con excepción de dos son significativas en nivel de p<0.05 y con excepción de ocho tienen una significancia de p<0.01. Sin embargo, los valores reflejan las relaciones demográficas conocidas entre las poblaciones, con el valor más bajo encontrado entre las muestras de Lucanas, y valores bajos en las poblaciones de Puno y el sur de Junín. Estos grupos poblacionales son claramente dependientes demográficamente de acuerdo a estos datos.
La dependencia o independencia demográfica puede también inferirse con los valores Nm mostrados debajo de la diagonal en la tabla 5. Generalmente debido a los altos niveles de FST encontrados en nuestro estudio, muchos valores de Nm son cercanos a la unidad. El valor Nm de uno indica que aproximadamente un migrante es intercambiado entre dos poblaciones de igual tamaño por generación (cada n años), lo que de acuerdo al trabajo pionero de Franklin y Soulé (1980) es suficiente para prevenir divergencia a través de “gene drift”. El límite de un migrante por generación (OMPG) ha sido subsecuentemente usado como regla para mantener la interdependencia demográfica en el manejo de poblaciones. Sin embargo, dado que las poblaciones estudiadas varían considerablemente en tamaño, y muchas de ellas demográficamente aisladas por cientos de generaciones, tales valores son insignificantes si no se ponen en el contexto de la historia poblacional conocida.
Por ejemplo, en términos biogeográficos es sorprendente que Yantac supuestamente ha intercambiado 2.7 migrantes por generación con Lucanas 1 mientras que solamente ha mantenido intercambio de 1.12 con su vecino Cachi Cachi. Estos resultados pueden ser parcialmente debido a los sesgos mencionados arriba, y deben interpretarse en el contexto de una historia poblacional conocida. En general, los valores Nm reflejan una historia demográfica aparente de la misma manera que los valores FST, aunque existen algunos valores anómalos, que serán discutidos posteriormente.
Se calcularon las distancias genéticas, porque estos aparentemente, reflejan la historia evolutiva de poblaciones de una manera más exacta que los índices demográficos como FST (Nei, 1987), y pueden ser más apropiadas para análisis por agrupamiento algoritmico, tal como “neighbour-joining” (agrupando vecinos) para producir dendogramas, los cuales pueden reflejar relaciones evolutivas entre las poblaciones estudiadas. Hemos usado valores pareados de distancias genéticas sin sesgo de Nei 1972 (valores no mostrados pero que correlacionan ampliamente con FST) para producir el dendograma “neighbour-joining” (agrupando vecinos) mostrado en la figura 2.
Las longitudes de las ramas son proporcionales a las distancias genéticas pero no indican escala en el tiempo evolutivo desde que los valores de la distancia genética pueden ser fuertemente influenciados por fluctuaciones demográficas. Sin embargo, los resultados nos permiten inferir relaciones entre poblaciones que tengan sentido demográfico y biogeográfico. Por ejemplo, dos de las tres muestras provenientes del norte y noroeste de Junín se agrupan muy cercanamente, lo mismo hacen las tres muestras del sur de Junín y las dos muestras de Puno. La estructura no es muy fuerte para las muestras de Lucanas, Arequipa y Huancavelica, sin embargo esto puede indicar una afiliación demográfica débil entre estas poblaciones que no es aparente en el análisis de distancia genética.
Ver Tabla
Finalmente, los datos fueron analizados usando análisis de correspondencia factorial bidimensional (2D-FCA) con la finalidad de aclarar las relaciones entre las poblaciones. FCA es un análisis canonical particularmente bien adaptado para describir relaciones entre dos variables cualitativas (aquí cero, uno o dos para cada alelo en cada locus) resultando en vectores de frecuencia alélica producidos para cada población y posteriormente analizadas usando el estimador FST de Robertson y Hill (1984) para producir una serie de valores inerciales para cada estimado FST por cada locus por cada individuo.
Cada eje de los valores inerciales puede ser analizado en un espacio multidimensional para producir un plot de correspondencia factorial, el más preciso de lo cual representa la mayor proporción del total del valor inercial. El resultado es un método muy poderoso para recuperar la máxima información sobre relaciones genéticas de individuos dentro y entre poblaciones usando un espacio n-dimensional. Para los datos de las vicuñas, los primeros dos ejes explican aproximadamente 6.5% del valor total inercial, una proporción mayor que el promedio para análisis intraespecíficos y un reflejo de la relativa gran fuerza de los datos.
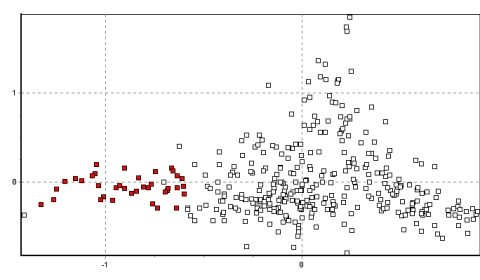
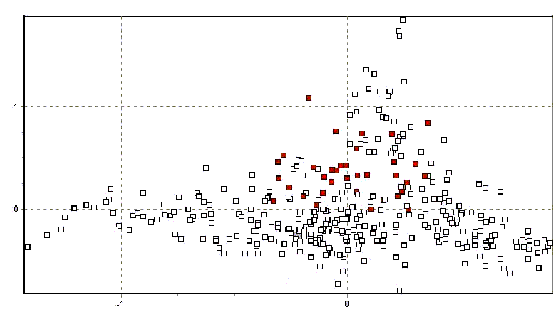
La figura 3 muestra los resultados de todos los datos en dos dimensiones. Es bien aparente el agrupamiento muy cercano de los individuos de cada población y las ubicaciones únicas que muchas de las poblaciones ocupan un espacio bidimensional. Especialmente notables son las poblaciones de Picotani (en azul oscuro: la única población encontrada entre vectores 0.6 y 1.2 en el eje x), y Yantac (en verde claro: la mayor población encontrada entre los vectores 0.9 y 1.5 en el eje y). En adición, varios grupos de poblaciones se hacen aparentes debido a su fuerte agrupamiento en el espacio 2D. Se identifican cuatro grupos en el ploteo, y las figuras 4a-d resaltan lo mencionado y explican su origen.
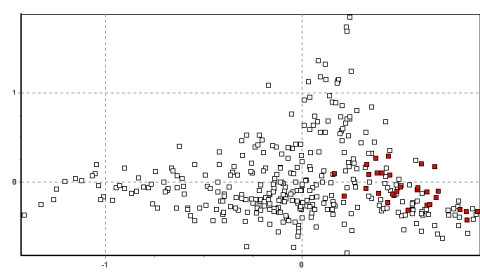
Primeramente, las poblaciones de Yantac, Villa Junín y en menor grado Tinco Cancha en el noroeste y norte de Junín forman un grupo, ocupando (x/y) 0-0.5/0.2-1.5 (ver Figura 4a) De estos, tres, Yantac es claramente el más diferente y ocupa la posición más noroeste, Villa Junín ocupa un lugar intermedio en el espacio bidimensional relativo a las muestras del sur de Junín. Tinco Cancha, sin embargo tiene una distribución más central, similar a las muestras de Lucanas (ver Poblaciones Anómalas abajo).
Las poblaciones en el sur de Junín se agrupan muy cercanamente, ocupando predominantemente los rangos coordinados de 0-0.5/-0.5-0 en el vector (x/y) la sección de abajo de la mano derecha del plot (ver Figura 4b). Este espacio coordinado es también único entre poblaciones de vicuñas peruanas y es un fuerte indicio de una historia demográfica independiente en esta región.
La principal característica del plot 2D-FCA es un gran grupo de poblaciones en el centro del vector de distribución. Agrupa todas las muestras de Lucanas, Ayavi, Huacarpana, Toccra y en menor grado Ingenio en Puno (rango x/y -0.5 a 0.2/-0.8 a 0.5). Estos resultados recapitulan el dendograma de distancia genética (Figura 2), y sugiere que las poblaciones desde Huancavelica hasta Ayacucho y Arequipa forman un solo grupo demográficamente unido de subpoblaciones. También existe evidencia de conexión con las poblaciones de Ingenio en el lado sur del Lago Titicaca en Puno. Es interesante anotar la fuerte relación entre la población de Huacarpana (ver Poblaciones Anómalas abajo) y Lucanas (también evidente en los datos de FST) y el hecho que los datos de Toccra deben ser tomados con precaución debido al poco número de muestras.
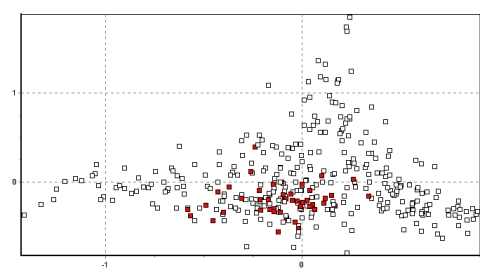
Finalmente la población de Picotani, Puno observada en la Figura 4d ocupa su propio espacio 2D (x/y= -1.5 a -0.5/-0.8 a 0.2) y es muy distinta. Situado al Norte del Lago Titicaca, Picotani no está lejos de la frontera con Bolivia. Su extrema diferencia (en escala similar a Yantac) claramente requiere atención particular.
Aunque la gran mayoría de las poblaciones de vicuñas estudiadas forman parte de uno de los cuatro grupos demográficos (ver arriba) existen algunas excepciones. Primeramente, la población de Tinco Cancha en el norte de Junín se parece genéticamente al grupo Andes centrales mucho más que al grupo de Junín septentrional (un efecto similar es evidente pero menos extensivo en Villa Junín)(Figura 5) y segundo, Huacarpana en Ica, contrariamente a lo esperado, es genéticamente más similar a las poblaciones de Lucanas. Documentos históricos sin embargo, hace evidente que estas poblaciones han sido afectadas por dos traslados provenientes de la Reserva de Pampa Galeras (Lucanas)(ver 1 y 3 en Tabla 6). Futuras estrategias de manejo deberán tener esto en cuenta, debido a que los traslados pueden afectar severamente la estructura genética de poblaciones locales cuando ello ocurre. En tabla 6, detallamos información sobre los traslados de vicuñas realizados en tiempos recientes. Hemos documentado el impacto de traslados 1 y 3 en el genoma de poblaciones locales, pero falta estudiar la influencia de traslados 2, 4 y 6.
CONCLUSIONES Y RECOMENDACIONES DE CONSERVACION
Las poblaciones de vicuñas en el Perú parecen poseer interesantes y fuertes características genéticas, las cuales son el resultado de su biología, el hábitat que ocupan, su historia evolutiva y por el manejo humano en el pasado reciente.
Primero, la diversidad genética de la vicuña peruana se caracteriza por los relativamente bajos niveles de diversidad genética dentro de las poblaciones, pero altos niveles de diferenciación genética entre poblaciones. Estos patrones son comúnmente observados en especies amenazadas que originalmente habitaban una gran extensión geográfica y posteriormente se aislaron unas de otras, sufriendo una drástica reducción demográfica en generaciones recientes (ej. búfalo sudafricano, O’Ryan et al., 1998 ; ardillas rojas, Barratt et al. 1999). Las características genéticas observadas en las vicuñas peruanas parecen ser el resultado de un proceso similar y deben ser tomadas en cuenta en el futuro desarrollo de estrategias de manejo, las que deben ser diseñadas para minimizar adicionales pérdidas de diversidad genética dentro de poblaciones individuales de vicuñas.
Es especialmente importante que los futuros planes de manejo para producción de fibra en vicuña se tomen en cuenta la diversidad genética de las poblaciones y que los reproductores (especialmente machos) deben ser monitoreados y manejados para maximizar la diversidad genética. Es importante preveer que los individuos puedan desplazarse libremente y evitar, por ejemplo, el uso exclusivo de unos pocos machos reproductores o cualquier programa dirigido a mejorar la finura y/o otras características de la fibra. La intervención humana solo ocasionaría mayor consanguinidad y cambios genéticos (“genetic drift“) con efectos negativos en la salud de individuos en la población. Además, las estrategias descritas arriba no conducirían a un incremento en la calidad de la fibra desde que la domesticación en otras especies productoras de fibra han demostrado en todos los casos una reducción significativa de la calidad de la fibra. Finalmente, es importante anotar que la vicuña ha sido domesticada anteriormente, dando como resultado a la alpaca (Kadwell et al. en revisión).
Los altos números de alelos privados y la clara estructura biogeográfica de la diversidad genética en la vicuña, evidencian la influencia de factores adicionales mencionados en la determinación de los patrones. Evidentemente, las poblaciones de animales andinos ocupan uno de los más extremos y variables medioambientes topográficos del mundo, donde los drásticos cambios climáticos diurnos, trastornos geológicos y cambiantes rutas de colonización tendrían un rol crucial en la formación de las relaciones genéticas entre las poblaciones actuales. En este concepto, es razonable esperar que muchas poblaciones de vicuñas se han aislado naturalmente, reagruparon, y aislaron nuevamente a lo largo de su historia. Estos efectos, conjuntamente con la reciente influencia antropogénica en las poblaciones de vicuñas, hacen difícil estimar cuanto de los patrones genéticos mostrados en nuestros resultados se deben a una reciente explotación humana y cuanto es consecuencia natural de la historia evolutiva. Consecuentemente, cualquier propuesta para futuros manejos deberían prestar atención a los actuales patrones de diversidad genética descritos aquí, para evitar futuras pérdidas en diversidad y estructura.
Segundo, hemos identificado cuatro diferentes grupos demográficos : Junín noroeste, Junín sur, Andes centrales y Puno. Estos grupos podrían formar unidades de manejo separados para el futuro aumento demográfico dentro y entre las poblaciones de cada unidad. Debe anotarse que la población noroeste de Yantac es genéticamente única en este estudio y podría representar un grupo evolutivo septentrional que requiere especial atención e investigación futura. También se distingue en Puno la población de Picotani, y en menor grado Ingenio, que representarían un grupo genético posiblemente ligado a la población boliviana. Por lo tanto, también es importante establecer la relación genética entre la población de Picotani y la de Bolivia a fin de conocer hasta donde este grupo genético ha penetrado dentro de Perú. Las poblaciones meridionales de Junín forman claramente una unidad demográfica independiente digna de una especial consideración. Así mismo, la gran población central peruana Andina, incluyendo la altamente diversa población de Lucanas, al parecer forman una gran unidad de manejo demográfico. Sin embargo, debe advertirse que las muestras de Toccra analizadas aquí, no son los suficientes para arribar a conclusiones valederas.
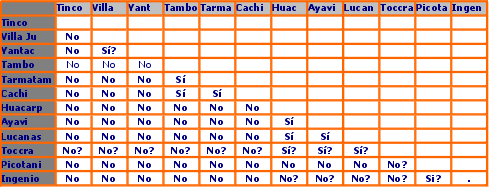
En la Figura 5 se muestra una matriz de manejo propuesta para cada par de poblaciones en este estudio conteniendo recomendaciones si las vicuñas pueden ser trasladas o no entre poblaciones según el esquema de manejo propuesto acá. Los signos de interrogación se deben al pequeño número de muestras de Toccra y a resultados ambiguos entre Ingenio - Picotani y Villa Junín - Yantac.
-
Las poblaciones de vicuñas en Perú deben manejarse cautelosamente como cuatro unidades demográficas: Junín noroeste (excepto Tinco Cancha), Junín sur, Perú central y Puno.
-
El manejo genético debe llevarse a cabo moviendo individuos entre poblaciones para prevenir adicionales pérdidas de diversidad genética.
-
desde Pampa Galeras a poblaciones fuera de la unidad de manejo del centro peruano, por el profundo efecto que podría tener en la estructura genética local (como ha ocurrido en Tinco Cancha).
-
El manejo genético dentro de poblaciones requiere medidas para minimizar los efectos de consanguinidad y cambios genéticos (“genetic drift”) . Esto debe tenerse en cuenta en las prácticas de manejo para producción de fibra. Debe asegurarse el libre movimiento de los individuos, y de existir barreras que impidan el libre movimiento (ej. cercas) deberán mantenerse en forma permanente grandes espacios abiertos, los que pueden cerrar solamente para prácticas de manejo intensivo (ej. chacu). Desde que el uso de cercos actúan como barreras para el libre movimiento de animales, se deberá buscar métodos de manejo intensivo que sustituyen las cercas a mediano plazo.
-
Debe darse prioridad a obtener muestras para el análisis genético de la región de Ancash, para ver si estas poblaciones comparten el mismo genotipo septentrional como el ejemplificado en Yantac.
-
Deben también obtenerse muestras de Toccra (Aguada Blanca) para aumentar el número que se tiene actualmente.
-
También deben obtenerse muestras para futuras investigaciones del grupo septentrional de Puno, las muestras deben comprender Bolivia así como del norte y oeste de Picotani, para establecer la penetración de este grupo en el Perú.
-
Así mismo debe aclarar la relación entre las poblaciones de vicuñas ubicadas al norte y sur del Lago Titicaca mediante la obtención de más muestras de la zona sur.
-
Deben realizarse análisis fenotípico (incluyendo fibra) de las cuatro subpoblaciones identificadas .
Se debe elaborar una versión simplificada y corta de este reporte para su distribución entre las partes interesadas incluyendo campesinos.
-
Se recomienda un estudio similar en poblaciones de guanaco en el Perú.
Los autores agradecen la ayuda de la Dra. Helen Stanley del Instituto de Zoología de Londres en el diseño del proyecto. Agradecen también a Valerie Richardson y María Stevens del Secretariado de la Iniciativa Darwin, DETR, Londres por su flexibilidad y apoyo en el manejo del proyecto. Al Instituto de IVITA, UNMSM en las personas de Dr. Felipe San Martín, Ing. Juan Olazabal y Antonina Cano. A CONACS en las personas de Dr. Alfonso Martínez, Jorge Herrera, Marco Antonio Zuñiga, Marco Antonio Escobar, Alex Montufar, Roberto Bombilla y todas las demás personas que ayudaron en la obtención de muestras.
-
EM Barratt, J Gurnell, G Malarky, R Deaville y MW Bruford. 1999. Genetic Structure of fragmented populations of red squirrel (Sciurus vulgaris) in Britain. Mol. Ecol. S12:55-65.
-
Belkhir L. 1999. GENETIX v4.0. Belkhir Biosoft, Laboratoire des Genomes et Populations, Université Montpellier II.
-
Benzécri, JP. 1973. L'Analyse des Données: T. 2, I' Analyse des correspondances. Paris: Dunod.
-
Bruford MW, Hanotte O, Brookfield JFY y Burke T. 1998. Single and multilocus DNA fingerprinting. En: Molecular Genetic Analysis of Populations: A Practical Approach, 2nd Edition. A.R. Hoelzel (ed). Oxford University Press, Oxford.
-
Franklin I y Soulé M. 1980. Conservation and Evolution. Longman, New York.
-
Goldstein D y Schlötterer, C (eds.) 1999. Microsatellites: Evolution and Applications. Oxford University Press, Oxford.
-
Kadwell M, Fernández M, Stanley HF, Wheeler JC, Rosadio R y Bruford MW. 2000. Genetic analysis reveals the wild ancestors of the llama and alpaca. In review.
-
Kumar S, Tamura K y Nei M. 1993. MEGA: Molecular Evolutionary Genetics Analysis, version 1.01. The Pennsylvania State University, University Park, PA.
-
Lang KDM, Wang Y y Plante Y. 1996. Fifteen polymorphic dinucleotide microsatellites in llamas and alpacas. Anim. Genet. 27:293.
-
Nei M. 1972. Genetic distance between populations. Am. Nat. 106:283-292.
-
Nei, M. 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
-
O’Ryan C, Harley EH, Bruford MW, Beaumont MA, Wayne RK y Cherry MI. 1998. Microsatellite analysis of genetic diversity in fragmented South African buffalo populations. Anim. Cons. 1:124-131.
-
Penedo MCT, Caetano AR y Cordova KI. 1998. Microsatellite markers for South American camelids. Anim. Genet. 29:411-412.
-
Raymond M y Rousset F. 1995. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86:248-249.
-
Roberston A y Hill WG. 1984. Deviations from Hardy Weinberg proportions: sampling variances and use in estimation of inbreeding coefficients. Genetics 107:703-718.
-
Saitou, N y Nei M. 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425.
|



{kind=link}
{kind=link}
{kind=link}